Dyzostoza żuchwowo-twarzowa po raz pierwszy została opisana już w 1900 roku przez chirurga i okulistę Edwarda Treachera Collinsa. Choroba dotyczy niedorozwoju kości policzkowych i kości szczęk, co objawia się pod postacią mnogich zaburzeń morfologicznych w obrębie głowy, ale i całego ciała. Stosuje się leczenie objawowe, ponieważ przyczynowe nie jest możliwe.

Dyzostoza żuchwowo-twarzowa – przyczyny
Schorzenie rozwija się na podłożu zaburzeń embriogenezy I i II łuku skrzelowego. Zwykle bezpośrednią przyczyną jest spontaniczna, niekontrolowana mutacja w genie TCOF1, zlokalizowanym na chromosomie 5 (5q32-q33.1). Do dnia dzisiejszego opisano około 116 różnych mutacji TCOF1, spośród których najczęstsze są delecje. Wciąż nie wiadomo jednak, co powoduje wystąpienie takich mutacji. Nieznane są nawet czynniki ryzyka.
Aż w 60% przypadków obserwuje się mutację spontaniczną, natomiast w pozostałych 40% dyzostoza żuchwowo-twarzowa występuje rodzinnie. Jest wtedy dziedziczona w sposób autosomalny dominujący.
Dyzostoza żuchwowa-twarzowa objawy
Ta rzadka choroba genetyczna objawia się przede wszystkim:
- małożuchwiem;
- zniekształconymi oczami w ustawieniu skośnym;
- rozszczepieniem powiek;
- brakiem policzków wskutek niedorozwoju kości jarzmowych;
- hipoplazją małżowin usznych i ucha wewnętrznego, co może powodować niedosłuch i głuchotę;
- rozszczepem podniebienia;
- zniekształceniem nosa;
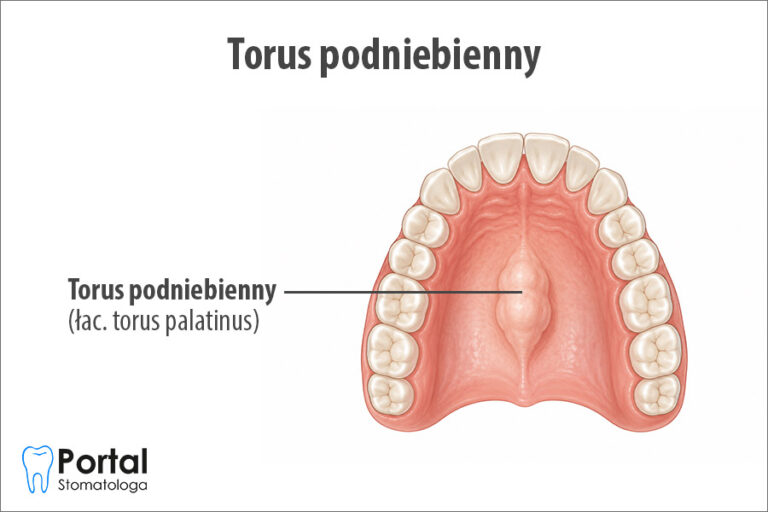
- podniebieniem gotyckim;
- zanikiem nozdrzy tylnych;
- zębami nadliczbowymi;
- zmętnieniem szkliwa;
- mikrodoncją;
- brakiem zawiązków zębowych.
Wady te przyczyniają się do znacznych trudności z prawidłowym mówieniem, oddychaniem, a nawet ze spożywaniem stałych pokarmów.Co istotne, rozwój intelektualny i emocjonalny dziecka są prawidłowe, dlatego dyzostoza żuchwowo-twarzowa jest dla nich szczególnie trudna. Wszelkie zaburzenia estetyki wyglądu przekładają się na poczucie własnej wartości oraz problemy natury psychicznej. W medycynie dyzostozę żuchwowo-twarzową dzieli się na 4 rodzaje:
- I – niewielki niedorozwój kości tworzących staw skroniowo-żuchwowy, przy czym czynność tego stawu zostaje zachowana. Częste jest tyłożuchwie i zgryz otwarty;
- II A – niedorozwój większy niż w klasie I, w efekcie czego staw skroniowo-żuchwowy może się przemieszczać;
- II B – ciężki niedorozwój kości tworzących staw skroniowo-żuchwowy. Pojawia się zaawansowane tyłożuchwie i zgryz otwarty;
- III – całkowity brak wyrostka kłykciowego i ramienia żuchwy, w efekcie czego żuchwa jest luźno ruchoma.
Ustalenie rodzaju ma znaczenie dla przyszłego leczenia.
Dyzostoza żuchwowo-twarzowa – diagnostyka
W diagnostyce najważniejsze jest wykonanie badań genetycznych, które z całkowitą pewnością potwierdzą obecność dyzostozy żuchwowo-twarzowej. Kluczowe są także badania obrazowe czaszki, ze szczególnym uwzględnieniem oceny stawów skroniowo-żuchwowych.
Dyzostoza żuchwowo-twarzowa – leczenie
Nie ma możliwości wyleczenia choroby, stosuje się więc leczenie objawowe. Dzieci już od najmłodszych lat muszą być objęte opieką stomatologiczną, okulistyczną oraz laryngologiczną. Zazwyczaj konieczne jest leczenie ortodontyczne, mające na celu korekcję wad zgryzu. Nierzadko podejmuje się czynności chirurgiczne przywracające symetrię i estetykę twarzy oraz korygujące wady, które mogą mieć wpływ na rozwój. Interwencja chirurgiczna konieczna jest choćby przy rozszczepach wargi i podniebienia.
Uzupełnieniem leczenia podstawowego jest fizjoterapia, która pozwala na właściwy rozwój ruchowy. Dzieci mogą potrzebować konsultacji psychologicznych lub psychoterapeutycznych, co jest jednak zawsze kwestią indywidualną, rozważaną osobno dla każdego dziecka z dyzostozą żuchwowo-twarzową.
Polecane produkty:

|
Kolagen naturalny bioalgi
Kolagen do picia to naturalny produkt z opatentowaną formułą Peptiplus® hydrolizowanego kolagenu. Dzięki temu jest bardzo wysokiej wchłanialności. Wzmacnia zęby, kości, ... Zobacz więcej... |
Bibliografia
- Mączka G., Szeląg J., Zespół Treachera Collinsa – przegląd piśmiennictwa, Dental and Medical Problems, 3/2009.
- Wójcicki P., Marszałek-Kruk B., Uwarunkowania genetyczne oraz zasady leczenia zespołu Treachera-Collinsa, Dental and Medical Problems, 4/2005.
- Kot M., Lewandowska M., Kruk-Jeronim J., Rodzinne występowanie zespołu Treachera Collinsa – opis 6 rodzin, Pediatria i Medycyna Rodzinna, 5/2009.